
Sciences & Technology
Welcome to the mRNA revolution



Discovery of an inflammatory driver in a rare syndrome may have implications for more common diseases like Parkinson’s and viral infections

Published 27 February 2022
“How many people actually have this disease?”
It’s a potentially triggering question at a family BBQ. I had just finished explaining why I’d recently travelled to the US to work on samples from patients with a rare disease called Proteasome Related Autoinflammatory Syndrome (PRAAS). None of my family had ever heard of this disease, which isn’t surprising. There are less than one thousand patients diagnosed with PRAAS worldwide. So, why work on it?

Why not instead work on a neurodegenerative disease like Parkinson’s for which there are over 2,225 newly diagnosed cases every year in Australia alone? Or perhaps a chronic viral infection? In 2019 over 150,000 people in Australia were living with a chronic hepatitis B infection.
It is a good point and arguments can be made for prioritising investment where the most good can be done. But clearly, neither can we abandon the people and families suffering from rare diseases.
Sciences & Technology
Welcome to the mRNA revolution
It is encouraging then that in 2020 Australia launched its National Strategic Action Plan for Rare Diseases, which involves dedicating to rare diseases some funds from the Medical Research Future Fund and the National Health and Medical Research Council.
But also, it needs to be remembered that research on rare diseases contributes significant new knowledge and understanding not just for the potential benefit of the patients affected, but also for our fundamental understanding of how the body works and fights diseases generally.
Take for example, PRAAS. The clinical presentation of PRAAS involves repetitive fever, severe rashes, immobility in the joints, muscle wastage and delayed physical development. Until 2018 the prognosis for PRAAS was dire. While immunosuppressive drugs could alleviate some symptoms patients ultimately succumbed to respiratory or cardiac failure. But, in 2018 a new treatment was designed which stopped the spread of inflammation and patient outcomes have improved.
Like most rare diseases it’s caused by inborn genetic errors – in PRAAS these errors affect a cell component called the proteasome. Proteasomes are complexes in our cells that identify and destroy unwanted proteins that can otherwise build up and eventually kill the cell.

The problem in PRAAS patients is that genetic errors occur in the creation of proteasome complexes meaning the proteasome isn’t correctly assembled and cannot function.
As a result, dangerous levels of protein ‘trash’ accumulates in the cells of these patients. Interestingly, this induces an inflammatory response from the cell which looks akin to the inflammatory response cells have to a viral infection.

Health & Medicine
Cannibal immune cells could offer new treatment path
Now, this is very interesting, because in diseases like Parkinson’s it has long been observed that unwanted proteins accumulate in the cells of patients and it is becoming increasingly understood that inflammation contributes to Parkinson’s disease. Furthermore, viruses like hepatitis B have evolved mechanisms to obstruct our cell’s proteasome so that they can replicate inside us.
These observations and many others demonstrate that the accumulation of unwanted proteins can trigger an inflammatory response from a cell, however, the specific molecular mechanism of how this happens wasn’t known, until we looked at PRAAS.
Studying how the loss of function in the proteasome drives inflammation in something like Parkinson’s or chronic viral infection is incredibly difficult because the cause and action of these diseases are complex, involving genetic and environmental factors.
In contrast, PRAAS has a defined genetic mutation that results in the specific loss of proteasome function, and this directly leads to inflammation. These are dots we can connect.
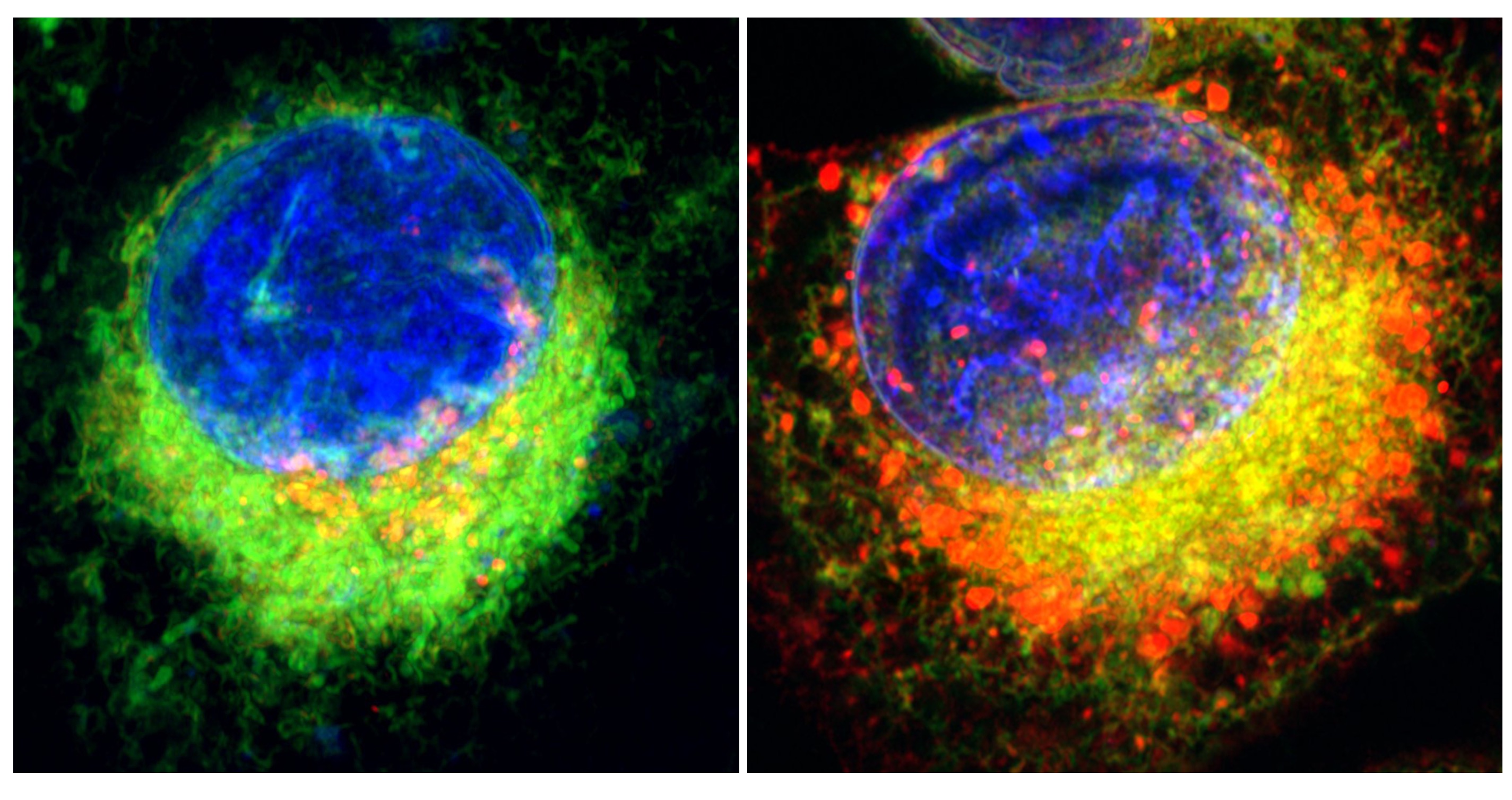
By using PRAAS as a system to study how our cells sense the build-up of protein trash, our research team found that a pattern-recognition receptor called Protein Kinase R (PKR) recognises the accumulation of a certain unwanted protein called IL-24.
Now IL-24 normally functions outside the cell so it can only accumulate inside the cell when the proteasome is not there to clear it away. If IL-24 isn’t cleared away by the proteasome, it is sensed by PKR which then triggers an inflammatory response.
Health & Medicine
Premature birth linked to lung disease later in life
Intriguingly, PKR is different from many other pattern recognition receptors. Along with driving inflammation it also activates a secondary pathway to inhibit the generation of new proteins by the cell. When a cell is stressed, generating new proteins can increase the accumulation of protein trash.
The action of PKR is therefore twofold – it drives inflammation to alert cells in the surrounding environment to a possible infection and it also attempts to reduce the burden of protein trash on the proteasome, hopefully restoring the cell to normal.
Inflammation is important for clearing viral infections, so activation of this pathway can protect cells from infection. However, when a cell cannot return to normal due to inborn genetic errors in a disease like PRAAS, PKR activation can lead to chronic inflammatory disease.
By studying PRAAS we were able to find a previously unknown link between inflammation and the build-up of unwanted proteins. This identified PKR as a potential target for new therapeutics aimed at inhibiting chronic inflammation in PRAAS.

Although it was discovered in a rare disease, this pathway may have implications in neurodegenerative diseases like Parkinson’s where protein build-up, inflammation and activation of PKR have all been observed but not yet functionally associated.
Furthermore, it may have implications for combating viral infections since it is possible that this particular inflammatory pathway evolved to combat viral infection.
Understanding this pathway and gaining these insights was only possible because PRAAS has a known genetic cause.

Health & Medicine
Explaining AI’s role in precision medicine
In Australia, a disease is considered rare if it affects less than five in 10,000 people. Although the term ‘rare’ suggests not many people are affected with a condition, there are over 7,000 rare diseases and this means collectively, around 8 per cent of Australians, or two million people, live with a rare disease.
Sadly, diseases like PRAAS are life-threatening or chronically debilitating, and as they are generally caused by inborn genetic errors, the majority of patients are children.
These two million Australians living with a rare chronic disease are actually helping us to further our understanding of human physiology and accelerate the development of treatments for thousands of diseases and conditions both common and rare. Their diseases deserve to be studied.
February 28 is Rare Disease Day, raising awareness and change for the 300 million people worldwide living with a rare disease, their families and carers.
Banner: Getty Images