
Health & Medicine
The genomic jigsaw of cancer


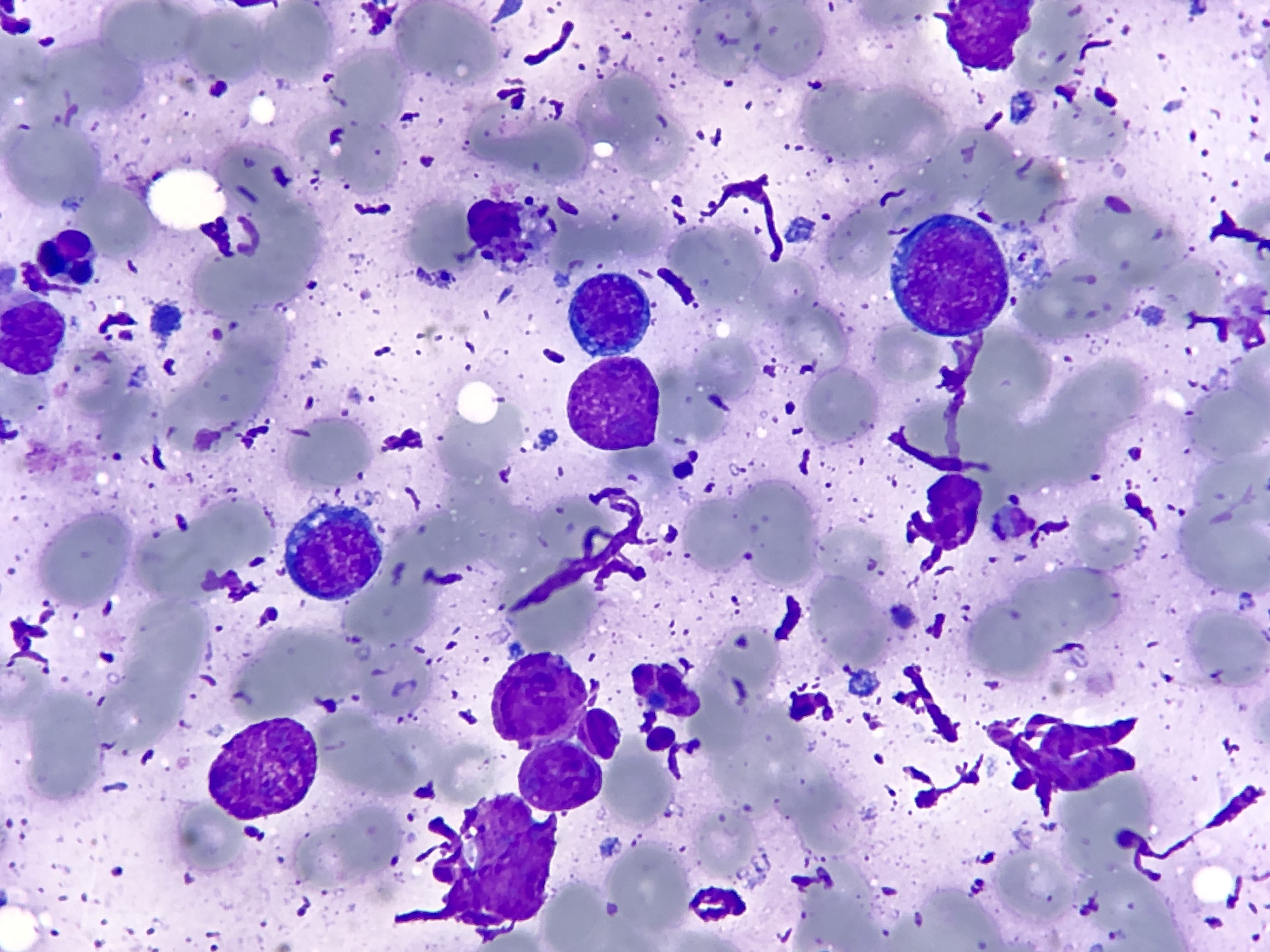
Recent genetic research has identified a surprising vulnerability of lymphoma cells that may lead to a new approach for cancer treatment

Published 20 February 2020
Cancer is a malady of our genes: certain DNA mutations that accumulate within a cell and its progeny enable them to wildly proliferate, overcoming the myriad of checkpoints that protect against unwarranted cellular growth.
Cancer-provoking (oncogenic) mutations fall into two major types: those that exaggerate the activity of normal growth-promoting genes (‘jamming the accelerator’) and those that remove the activity of normal growth-suppressing genes (‘destroying the brakes’).
Fortunately, a single oncogenic mutation is not sufficient to cause a cancer. Cancers only arise through the successive occurrence of multiple mutations that synergise to enhance cell production.
A gene known as MYC encodes a protein which activates (‘switches on’) numerous genes involved in promoting cell production.
Health & Medicine
The genomic jigsaw of cancer
In normal cells, the level of MYC protein is low and strictly regulated. But cancer cells have lost these safeguards and MYC levels are unchecked, provoking unbridled cell multiplication.
In the early 1980s, Professor Jerry Adams and I started studying MYC-driven cancers when our team discovered the molecular basis for human Burkitt’s lymphomas - a highly aggressive form of non-Hodgkin’s lymphoma.
This malignancy is rare in Australia, but quite common in central Africa and Papua New Guinea, particularly in children.
Burkitt’s lymphomas develop from antibody-producing B lymphocytes and every tumour had been found to carry a very distinctive chromosome abnormality.
Using gene cloning, we discovered that the chromosome abnormality results from genetic rearrangements that link the MYC gene to strong ‘on’ switches found next to antibody genes.

This discovery suggests that high levels of MYC foster malignancy. But this was merely ‘guilt by association’ – we needed direct proof.
We managed to obtain this proof by generating a mouse model of Burkitt’s lymphoma. Every mouse expressing the abnormal MYC gene in its B lymphocytes eventually developed lymphoma, proving that a high expression of MYC is the fundamental cause of Burkitt’s lymphoma.
It is now known that up to 70 per cent of human cancers have abnormally high levels of MYC protein.
Health & Medicine
The Global Cancer Atlas
Different cancer types overexpress MYC through a variety of genetic mistakes. Irrespective, their common feature of high MYC is a fundamental driver. And this, in turn, implies a common vulnerability.
Over the last 20 years, there has been huge progress in the outlook for cancer patients through the use of new medicines which specifically target proteins promoting cancer development and progression.
For many years, we hoped for a drug which could directly target the MYC protein as a potential cancer treatment.
However, the MYC inhibitors which have so far been developed have had disappointing results in clinical trials. It became clear we needed to look for other vulnerabilities in MYC-driven cancers.
One of these vulnerabilities is encoded by MYC itself.
This is because, as well as driving the expression of many cell division genes, MYC has its own ‘safety switch’ – MYC can activate the cell death programme known as apoptosis.
This means cells with high levels of MYC are very sensitive to cell death stimuli, unless they also contain high levels of proteins that inhibit apoptosis – such as the protein BCL-2 – which is itself upregulated in many cancers.
Our bodies are indeed a finely tuned system.

Health & Medicine
Big data puts genomic handbrake on cancer
Our search for targets to tackle MYC-driven cancers has taken many directions (including targeting BCL-2 itself).
Most recently, however, we homed in on the role of a protein related to MYC, called MNT.
MNT can target many of the same genes as MYC but instead of activating the expression of these target genes, it switches them off.
Our research, published in the journal Blood, found that deleting the MNT gene from lymphocytes expressing high levels of MYC protein greatly inhibits lymphoma development.
This may sound counterintuitive. But it turns out the most important aspect of MNT function, at least in lymphocytes, is to suppress MYC’s tendency to drive apoptosis.
This means that in the absence of MNT, lymphocytes with high levels of MYC drive themselves to death.
Dr Hai Vu Nguyen and Dr Cassandra Vandenberg discovered this using our laboratory model of human Burkitt’s lymphoma.
In this model, young mice have abnormally high numbers of MYC-driven B lymphoid cells. However, in mice lacking the MNT gene, they found startlingly low numbers of these abnormal-but-not-yet-malignant cells.
Health & Medicine
Tapping into the power of unusual white blood cells
Upon closer analysis, Dr Nguyen realised the MYC-driven B lymphoid cells were killing themselves.
He found that the cells had elevated levels of the protein BIM, one of the most important triggers of apoptosis. Tellingly, when he reduced the level of BIM, he found that the number of MNT-null/MYC-high lymphocytes was largely restored.
These studies established that MNT is required to keep MYC-driven cells alive, primarily by suppressing expression of BIM to reduce the risk of MYC-driven apoptosis.
But they were performed in premalignant cells. Do fully malignant cells, which have acquired numerous other oncogenic mutations, retain this sensitivity?
Using a genetic trick, our team went on to delete the MNT gene from fully malignant MYC-driven lymphomas and found that the tumour cells indeed died very rapidly.
This unexpected discovery totally reverses given wisdom about tackling MYC-driven cancers. Rather than inhibiting MYC, we should retain its activity and instead inhibit MNT, to maximise MYC’s innate ability to drive apoptosis.
MNT may also prove vital for the survival of other types of blood and solid cancer cells known to be driven by MYC and we have begun to test this.

Health & Medicine
Gene genies: Meet the researchers mapping our DNA to combat cancer
Our studies suggest that the expanding panel of new cancer therapeutics should include an effective MNT inhibitor.
As far as we are aware, no such inhibitor yet exists. But we hope that our findings will inspire scientists to vigorously pursue a MNT-inhibiting drug, perhaps even using Australia’s own National Drug Discovery Centre.
We anticipate that a drug that substantially inhibits MNT should not only elevate spontaneous apoptosis in MYC-driven tumours, but also increase their sensitivity to other cytotoxic drugs, most of which rely on triggering apoptosis.
After 40 years of research into MYC, I certainly hope that we have finally exposed its Achilles’ heel, and that cancer patients will one day benefit from these efforts.
The research was supported by the Australian National Health and Medical Research Council, the US-based Leukemia and Lymphoma Society, philanthropic support and infrastructure funding to the Walter and Eliza Hall Institute from the Commonwealth and Victorian Governments.
Banner: Burkitt Lymphoma/Ed Uthamn, Flickr


