


Sciences & Technology
The tip of the CRISPR iceberg


Seeing our invisible DNA architecture reveals that our genome is much more than a linear code, but rather an ever-changing blueprint
Published 14 December 2021
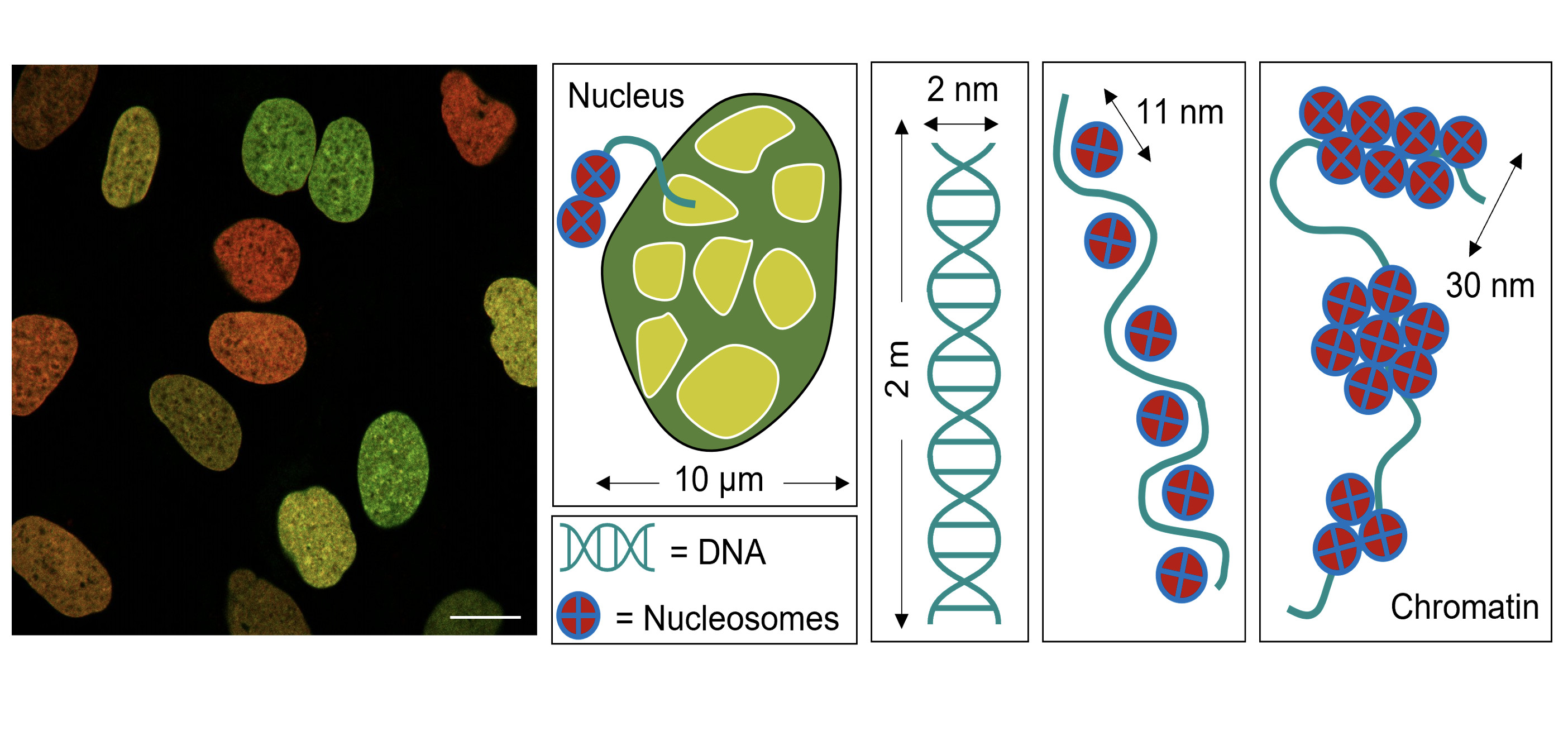
Inside the nucleus of a human cell, there are approximately two metres of DNA folded into a multi-layered 3D structure called chromatin, which allows all of our genetic information to be compacted into a tiny little space.
This 3D network of DNA is our genome and, intriguingly, whole genome sequencing has revealed that only two per cent of it is made up of genes that code the proteins that allow us to live, grow and reproduce.
The other 98 per cent – called noncoding DNA – used to be thought of as ‘junk DNA’. But it’s anything but junk.
Although we still don’t know the function of a large proportion of our noncoding genome, there is increasing evidence that it’s somehow involved in orchestrating the dynamic structural rearrangements in DNA that turn different protein-coding genes on and off.
DNA is made up of a series of molecules, designated by the letters A, C, G and T. Part of the sequence might look like this: ATCGGTGACTATCG. Within the billions of letters in this code, we can find and ‘read’ the genes to see what proteins they make.
Sciences & Technology
The tip of the CRISPR iceberg
But for the other 98 per cent of our genome, we need to look beyond the linear DNA template and figure out how our genome works in a 3D cell nucleus.
Directly observing whole genome dynamics in a living cell, and the local rearrangements in DNA that we think regulate gene expression is an immense challenge. This is because DNA is very small and the changes are so subtle that they are invisible to optical microscopy.
While the invention of optical microscopy and the use of spherical lenses to focus light has undoubtedly revolutionised our understanding of cell biology by allowing us to view things magnified hundreds of times, we can’t keep magnifying a biological specimen forever.
There is a limit to what spatial detail can be resolved, and this limit is caused by a phenomenon known as diffraction.
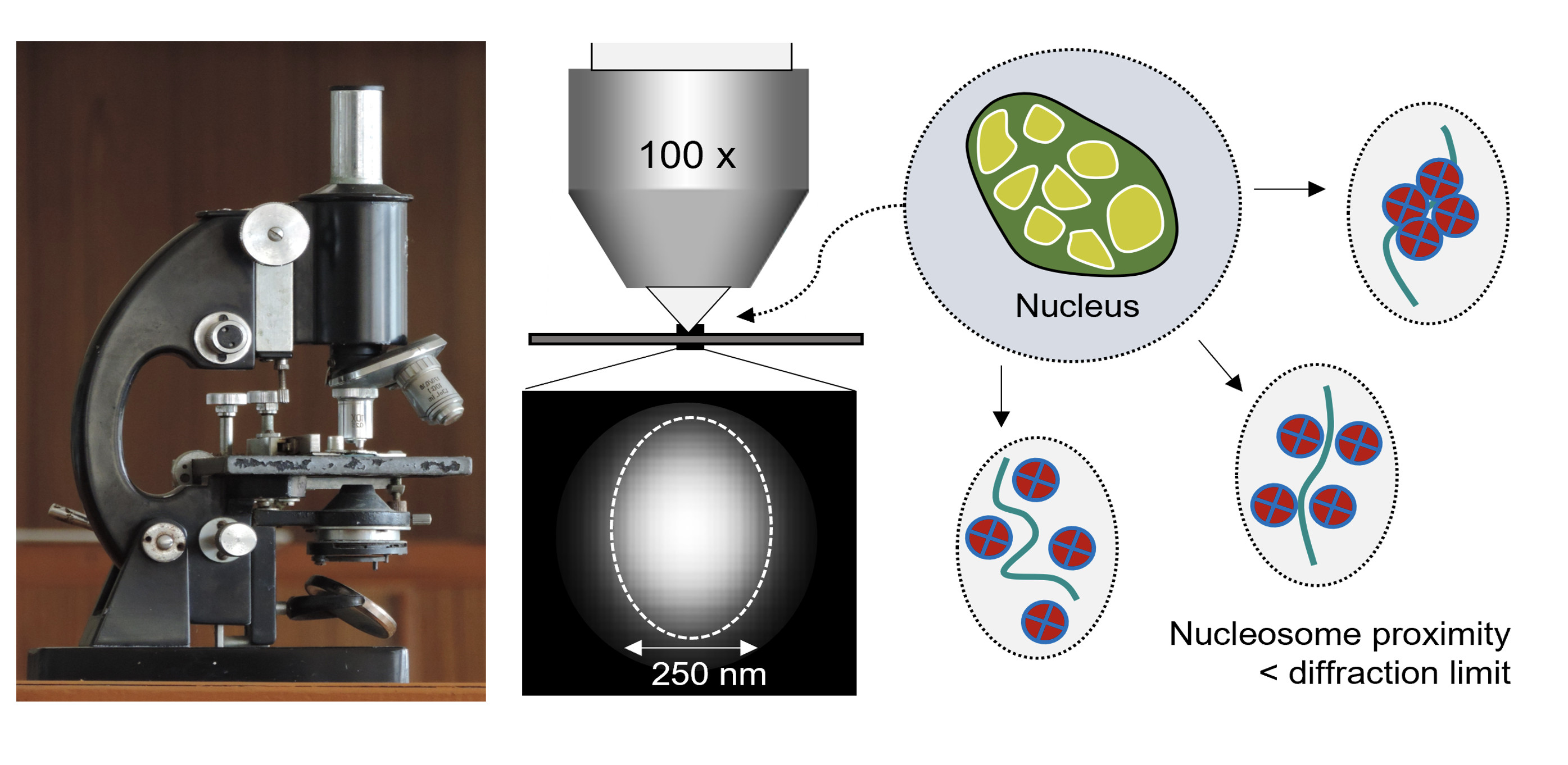
Because light travels in waves, the light ‘rays’ focused by the lens of an optical microscope do not actually converge into a sharp ‘point’ but instead form a blurry focal ‘spot’ with a width approximately equal to one-half of the wavelength of light involved (that is, 250 nanometres for green light).
Although that’s really small, it’s not small enough to see how the DNA template folds and unfolds in the nucleus of a living cell.
In particular, although lens-based microscopes can provide a sort of ‘bird’s eye view’ of changes in DNA structure, the nanoscale features of genome structure that are thought to regulate genome function are too small to see.
This ‘invisible’ but critical nanoscale feature is the spacing between subunits called nucleosomes and they are essentially genomic beads of DNA spooled around a core of proteins called histones.
Nucleosomes were first identified by electron microscopy which uses a beam of electrons (instead of light) to get around the diffraction limit of optical microscopy.
But since this method requires the cells to be fixed in place, only static snapshots of nucleosome arrangement can be obtained. What we want to see are the real-time changes in nucleosome spacing that expose different parts of our genome in a living cell.
Now we’re in an exciting phase of optical microscopy where the diffraction limit of lens-based microscopes can either be ‘broken’ or avoided altogether by using super-resolved readouts of genome architecture that look inside the blurry spots, or pixels, within a diffraction-limited image.

The key is fluorescence.
Fluorescence is a phenomenon where a molecule called a fluorophore absorbs light and then emits it at a longer wavelength. For example, a fluorophore might absorb blue light and then emit green light or absorb green light and then emit red light.
Importantly, we can decorate our DNA with different coloured fluorophores, and by observing their fluorescence with special microscopes, we can see details way beyond the diffraction limit that we’ve lived with for the past few centuries.
Health & Medicine
Deciphering ‘cell talk’ to understand our evolution
These new fluorescence methods can provide super-resolved information on genome architecture, down to just a few nanometers. And the ability to track this architecture in real-time means seeing the DNA network dynamics – that noncoding DNA is proposed to regulate – is now within reach.
In our lab, we investigate the role 3D genome organisation and dynamics play in maintenance of live cell nucleus function by adding green and red fluorescent proteins to the histones that compact DNA into a string of beads.
We then look for a phenomenon called Förster resonance energy transfer (FRET).
FRET is where a donor fluorescent molecule, instead of releasing light, transfers its absorbed energy to a nearby acceptor fluorescent molecule. And because this phenomenon is exquisitely sensitive to the distance separating the donor and acceptor molecule on a scale of one to 10 nanometers – detection of FRET can serve as a kind of molecular ruler.
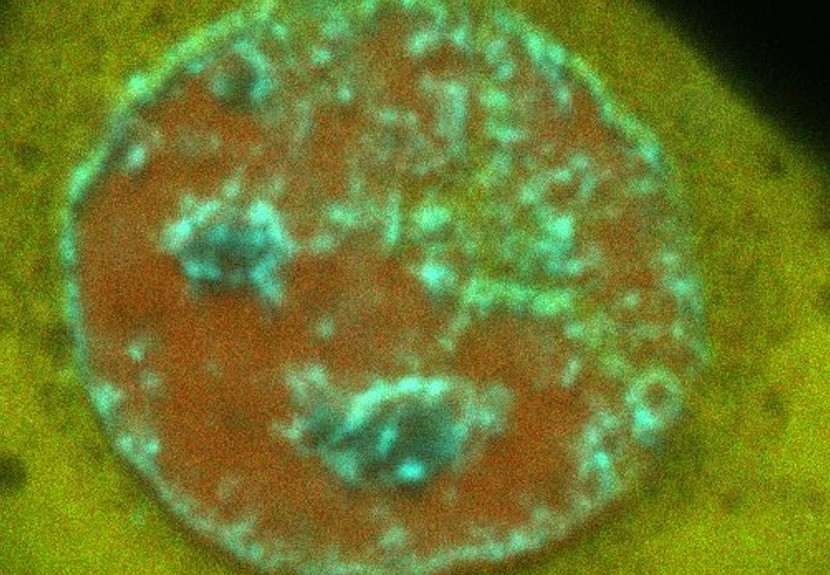
So, in a diffraction limited microscope image, by working out where fluorophores donate and accept energy, the nanoscale spacing between fluorescently labelled nucleosomes along the DNA template can be measured within each blurry spot that is approximately 250 nanometers wide.
This means being able to observe minute changes in the DNA architecture.
Using a fluorescence microscope specifically designed to spatially map these events, we have detected nanoscale rearrangements in DNA that ensure faithful transmission of our genome. In particular, for the first time ever, we have seen structural changes in DNA that promote the arrival of repair machinery at DNA damage sites.
Our work to date suggests chromatin architecture serves as a ‘road map’ for DNA-binding proteins to perform genome surveillance and localised DNA repair when damage arises.
Next time you hear the words ‘junk DNA’, remember it’s not junk. If you look much, much closer, it’s really an ever-changing blueprint for navigating the genome.
Banner: Live cell nuclei expressing a DNA repair protein accumulating at sites of damage (red)/ Supplied